


Sterke valentie-elektronen Afhankelijke en logische relaties van elementaire onzuiverheden in 2D binaire halfgeleider:een geval van GeP3-monolaag uit Ab Initio-onderzoeken
Abstract
Met behulp van eerste-principeberekeningen binnen de dichtheidsfunctionaaltheorie onderzoeken we de elektronische eigenschap en stabiliteit van substitutiegedoteerde 2D GeP3 monolaag met doteermiddelen van groep III tot VI. De geleidende eigenschappen blijken dramatisch te worden gewijzigd door zowel de doteringsplaatsen als het aantal valentie-elektronen van doteermiddelen. In het bijzonder vertoont substitutie op de Ge-site metaal-halfgeleideroscillaties als een functie van het aantal valentie-elektronen van doteermiddelen, terwijl dergelijke oscillaties volledig worden omgekeerd wanneer substitutie op de P-site. Bovendien bestuderen we ook het geval van co-doping in GeP3 , wat aantoont dat co-doping een logisch "EN"-fenomeen kan produceren, dat wil zeggen de geleidende eigenschappen van co-doped GeP3 kan worden afgeleid via een eenvoudige logische relatie volgens de resultaten van enkelvoudige doping. Ten slotte onderzoeken we de vormingsenergie van doteermiddelen en vinden dat de elektronen-gat en gat-gat co-gedoteerde systemen veel energetisch gunstiger zijn vanwege de Coulomb-aantrekking. Onze bevindingen bieden niet alleen een uitgebreid begrip van het 2D-dopingfenomeen, maar stellen ook een intrigerende route voor om de elektronische eigenschappen van 2D binaire halfgeleiders af te stemmen.
Inleiding
Sinds de ontdekking van grafeen [1, 2], de familie van tweedimensionale (2D) kristallen zoals overgangsmetaal dichalcogeniden (TMDs) [3], siliceen [4], germaneen [5], fosforeen [6], tellurene [ 7], enzovoort, hebben veel aandacht getrokken vanwege hun unieke elektrische, optische en magnetische eigenschappen [8,9,10]. Grafeen gedraagt zich bijvoorbeeld als massaloze Dirac-fermionen, wat leidt tot ultieme mobiliteit van ladingsdragers [11, 12]. Het is dus veelbelovend om het 2D-quantum-spin Hall-effect, verbeterde thermo-elektriciteit, supergeleiding [13] en zelfs het kwantum afwijkende Hall-effect [14,15,16] te ondersteunen. Gecombineerd met het groeiende aantal beschikbare kristalstructuurdatabases, zijn moderne computerhulpmiddelen gebruikt om nieuw onontdekte 2D-materialen te verkennen. Tot nu toe zijn er meer dan 1000 2D-materialen voorspeld en sommige ervan zijn gefabriceerd in experimenten [17,18,19], wat een interessant veld wordt in de fysische, chemische en materiaalwetenschap. Dergelijke fundamentele studies en verkenningen van 2D-materialen vergroten ook hun grote potentiële toepassingen in het detectieveld [20,21,22,23,24,25].
Onlangs hebben Jing et al. meldde nieuw 2D-materiaal-GeP3 monolaag, die een hogere chemische stabiliteit heeft dan BP-monolaag en uitstekende elektronische en optische eigenschappen bezit. Bovendien is de 2D GeP3 monolaag lijkt een halfgeleidende eigenschap te hebben vanwege de sterke kwantumopsluiting tussen de lagen. Ze ontdekten dat de GeP3 monolaag vertoont een gematigde en afstembare bandafstand van ongeveer 0,55 eV [26]. Op basis van de hoge capaciteit en goede cyclische stabiliteit, GeP3 dunne film wordt voorgesteld voor lithium-ionbatterijen als een veelbelovende anode [27]. Li et al. onderzocht ook de GeP3 nanoribbon en ontdekte dat de bandgaps even oneven oscillaties kunnen vertonen met de breedtetoename [28].
Doping is een praktische strategie om de elektronische en magnetische eigenschappen van de gastheer 2D-gelaagde materialen fundamenteel af te stemmen [29]. Bovendien doorbreekt het de beperking van een enkel materiaal in de toepassingen van vele velden en apparaten. Zoals we weten, kan de 2D-monolaagse halfgeleider resulteren in opmerkelijk verbeterde elektron-elektron-interacties waarvan is aangetoond dat ze renormalisatie en exciton met grote bandafstand genereren uit zowel theoretische berekeningen als experimenten met veel lichamen [30, 31]. Vergeleken met doping in bulkhalfgeleiders, wordt verwacht dat doping in 2D-halfgeleiders ook abnormaal gedrag vertoont vanwege het sterke elektronenopsluitingseffect, dat wil zeggen dat grafeen gedoteerd met boor of stikstof mogelijk is om een kleine bandgap op het Dirac-punt te openen, en de band gap van grafeen kan ook effectief worden geopend rond K (of K') punten door kleine BN-domeinen te introduceren [32]. Band gaps van zwart fosforeen vertonen een oscillerend gedrag door verschillende elementen te doteren met even of oneven aantallen valentie-elektronen [33, 34]. In dit werk proberen we het onderzoek naar dopingelementen van groep IV-V in 2D binaire GeP3 uit te breiden. monolaag halfgeleider.
Hier voerden we de systematische onderzoeken uit van het substituut-gedoteerde GeP3 monolaag met de doteermiddelen van groep III tot VI. De elektronische eigenschappen van gedoteerde systemen zullen dramatisch worden beïnvloed door zowel het aantal valentie-elektronen van doteermiddelen als de doteringsplaatsen. De centrale korrels zijn (1) voor enkelvoudig doteringsmiddel, de resultaten zijn gevoelig afhankelijk van de substitutieplaatsen en de substitutie op twee soorten doteringsplaatsen zal totaal omgekeerde resultaten opleveren. (2) De geleidende eigenschappen van co-doping kunnen door een logische operator worden afgeleid uit die van de enkele doteringsstof. Bovendien suggereert de berekende vormingsenergie van verschillende soorten doping dat sommige ervan zeer energetisch gunstig zijn tegen thermische fluctuaties.
Berekeningsmethoden
Al onze berekeningen van de dichtheidsfunctionaaltheorie binnen de algemene gradiëntbenadering worden uitgevoerd met behulp van Vienna ab initio Simulation Package [35]. De uitwisselings- en correlatietermen werden beschreven met de Perdew-Burke-Ernzerhof (PBE) -functionaliteit, en de projector versterkte golfpotentiaal werd gebruikt om de elektron-ion-interactie te beschrijven [36,37,38]. De gedoteerde GeP3 monolaag werd gemodelleerd in een periodieke 2 × 2 supercel met 32 atomen, en een grotere supercel van 3 × 3 werd ook gebruikt om onze resultaten te controleren. Een vacuümruimte van ongeveer 20 Å langs de z richting werd aangenomen om de interactie tussen aangrenzende lagen te elimineren. Voor de enkelvoudige dotering werd één Ge- of P-atoom vervangen door een doteringsstof uit groep III (IV, V en VI). De geometrische structuren worden bepaald door vergelijking met de gerapporteerde resultaten, inclusief roosterconstante en elektronische eigenschap van gastheer GeP3 monolaag. In de dopingsystemen mogen alle atomen in de supercellen ontspannen totdat de Hellmann-Feynman-kracht minder is dan 0,02 eVÅ −1 , maar de roosterconstanten van de oppervlaktecellen zijn gefixeerd tijdens de atoomrelaxatie. Een kinetische energiegrens van ongeveer 600 eV en 6 × 6 × 1 k -meshes werden gebruikt, respectievelijk [39].
Om de beschikbaarheid van de doteermiddelen in de GeP3 . te controleren monolaag, de vormingsenergie (E f ) van doteerstoffen X (X =groep III–VI) wordt berekend volgens de twee volgende formules. Voor enkelvoudige dotering hebben we het volgende:
$$ {\mathrm{E}}_{\mathrm{f}}\left(\mathrm{Ge}{\mathrm{P}}_3:\mathrm{X}\right)=\mathrm{E}\left (\mathrm{Ge}{\mathrm{P}}_3:\mathrm{X}\right)-\mathrm{E}\left(\mathrm{Ge}{\mathrm{P}}_3\right)-{ E}_{\mathrm{X}}+{E}_{\mathrm{i}} $$ (1)en voor het co-dopingsysteem wordt een vergelijkbare formule gebruikt:
$$ {\mathrm{E}}_{\mathrm{f}}\left(\mathrm{Ge}{\mathrm{P}}_3:\mathrm{XY}\right)=\mathrm{E}\left (\mathrm{Ge}{\mathrm{P}}_3:\mathrm{XY}\right)-\mathrm{E}\left(\mathrm{Ge}{\mathrm{P}}_3\right)-{ E}_{\mathrm{X}}-{E}_{\mathrm{Y}}+{E}_{\mathrm{i}}+{E}_{\mathrm{j}} $$ (2 )waar E f (GeP3 : X ) en E (GeP3 ) zijn de totale energieën van de X-gedoteerde en intrinsieke GeP3 monolaag met dezelfde supercel. E (GeP3 : XY ) is de totale energie van het gecombineerde XY-systeem, E X en E J zijn de atoomenergieën van doteermiddelen X of Y gerefereerd aan hun overeenkomstige bulkstructuren, en E ik , E j zijn de energieën van gesubstitueerde atomen waarbij i en j respectievelijk het Ge- of P-atoom aangeven [40, 41].
Resultaten en discussies
Even-oneven oscillaties voor enkelvoudige dopingsystemen
Figuur 1a toont het boven- en zijaanzicht van de structuur van de GeP3 2 × 2 supercel, en Fig. 1b is de overeenkomstige 2D Brillouin-zone van GeP3 monolaag. De geoptimaliseerde roosterconstanten van GeP3 monolaag zijn \( \mathrm{a}=\mathrm{b}=6.96\ {\AA} \), en de berekende band gap is ongeveer 0,26 eV, wat goed overeenkomt met andere theoretische berekeningen.
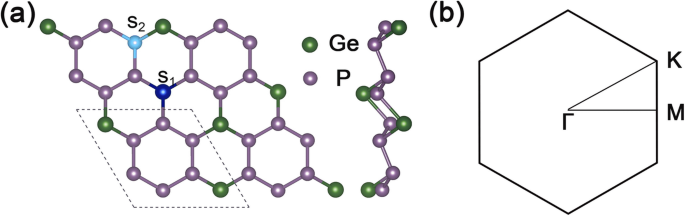
Geometrische structuur en Brillouin-zone van GeP3 . een Boven- en zijaanzichten van de geoptimaliseerde geometrie van GeP3 met een 2 × 2 supercel. De stippellijn geeft de eenheidscel van GeP3 . weer monolaag, S1 vertegenwoordigt de plaats van vervangingspositie van Ge-plaats, en S2 staat voor de plaats van het substitueren van positie P-atoom. b de 2D Brillouin-zone van GeP3 monolaag
Ten eerste hebben we de bandstructuren geplot van met één element gedoteerd GeP3 monolaag met een vervangend Ge-atoom (hier kiezen we B, C, N, O, Al, Si, P, S, Ga, As en Se als doteermiddelen). De resultaten worden respectievelijk getoond in Fig. 2a–l. We kunnen duidelijk zien dat het Fermi-niveau omhoog schuift en de geleidingsbanden kruist voor groep V (N, P, As) vanwege nog een elektrondotering, terwijl voor groep III (B, Al, Ga) doteringen vanwege één minder elektron, de men schuift naar beneden en passeert de valentiebanden. In Fig. 2f en j bijvoorbeeld, komt hun valentiebandmaximum net overeen met de gedeeltelijk bezette banden getoond in Fig. 2e en i. Voor groep IV (C, Si en Ge) en VI (O, S en Se) doteermiddelen vertonen de systemen echter een halfgeleiderfunctie, vanwege dezelfde of twee meer elektronen als het Ge-atoom. Een dergelijke afstemming van de overgang van halfgeleidende naar metalen komt voort uit de bezetting van het aantal valentie-elektronen, namelijk de bezetting van oneven (even) valentie-elektronen leidt tot metallische (halfgeleidende) eigenschappen.
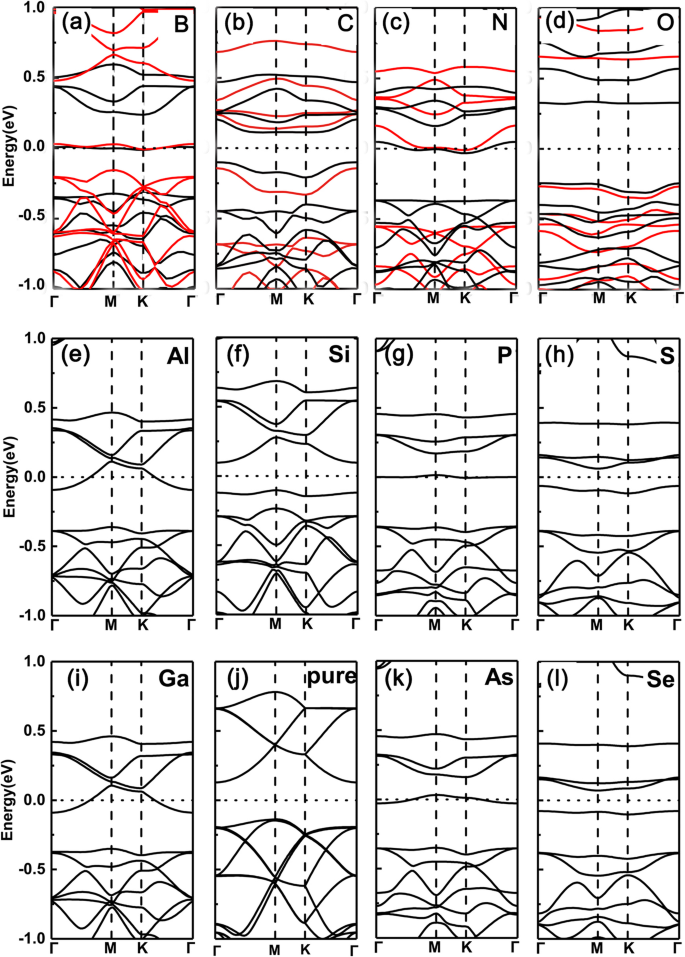
Bandstructuren van de verschillende doteermiddelen in GeP3 monolaag met vervangend Ge-atoom. een B, b C, c N, d O, e Al, f Si, g P, u S, ik Ga, j Pure GeP3 , k Als, l zie. Berekende bandstructuren voor een \( \mathsf{2}\times \mathsf{2} \) supercel met verschillende doteringen in GeP3 monolaag van groep III tot VI, waarbij respectievelijk het Ge-atoom wordt gesubstitueerd, samen met dat van zuiver GeP3 monolaag. Zowel de PBE- als de HSE06-functionaliteit worden gebruikt in de bovenste rij
Om de validiteit van de bovenstaande resultaten die zijn afgeleid van PBE-functionals te bevestigen, gebruiken we ook de hybrid density functionals (HSE06) functionalen om de gedoteerde systemen van de bovenste rij te controleren. Het is duidelijk dat PBE-functionaliteiten inderdaad de fouten van bandgaps geven vanwege de onderschatting. In onze bestudeerde systemen hebben ze echter allemaal aanzienlijke hiaten, dit betekent dat de fouten tussen metallische of halfgeleidende eigenschappen veroorzaakt door de PBE-functionalen meestal niet zullen optreden (dit komt omdat in sommige kleine bandhiaten van halfgeleiders PBE-functionalen meestal aanleiding geven tot tot de fout tussen geleidende en metallische eigenschappen). Bovendien gaat het ons in onze studie om de metallische of halfgeleidende eigenschappen, in plaats van de specifieke waarden van bandhiaten. Vergeleken met de hiaten die zijn afgeleid van PBE-functionalen, worden de hiaten van HSE06-functionalen duidelijk groter. Zelfs met deze blijven de metaal-halfgeleideroscillaties intact. Daarom zijn de centrale ingrediënten die zijn opgesteld op basis van PBE-functionaliteiten betrouwbaar.
In scherp contrast zijn de gevallen van vervanging van P-atomen door dezelfde doteermiddelen echter volledig omgekeerd, zoals respectievelijk getoond in Fig. 3a-l. Dat wil zeggen, voor groep V (N, As) en groep III (B, Al, Ga) doteermiddelen, blijven de gedoteerde systemen halfgeleidende eigenschappen, terwijl voor groep IV (C, Si, Ge) en VI (O, S, Se) doteerstoffen, die veranderen in een metalen functie (hier wordt dezelfde trend ook gevonden tussen PBE- en HSE06-functionaliteiten). Dit komt omdat de valentie-elektronen dezelfde (twee minder) behouden als (dan) de intrinsieke GeP3 voor groep V (groep III) doteerstoffen, maar één (meer) elektron minder voor groep IV (VI) doteerstoffen.
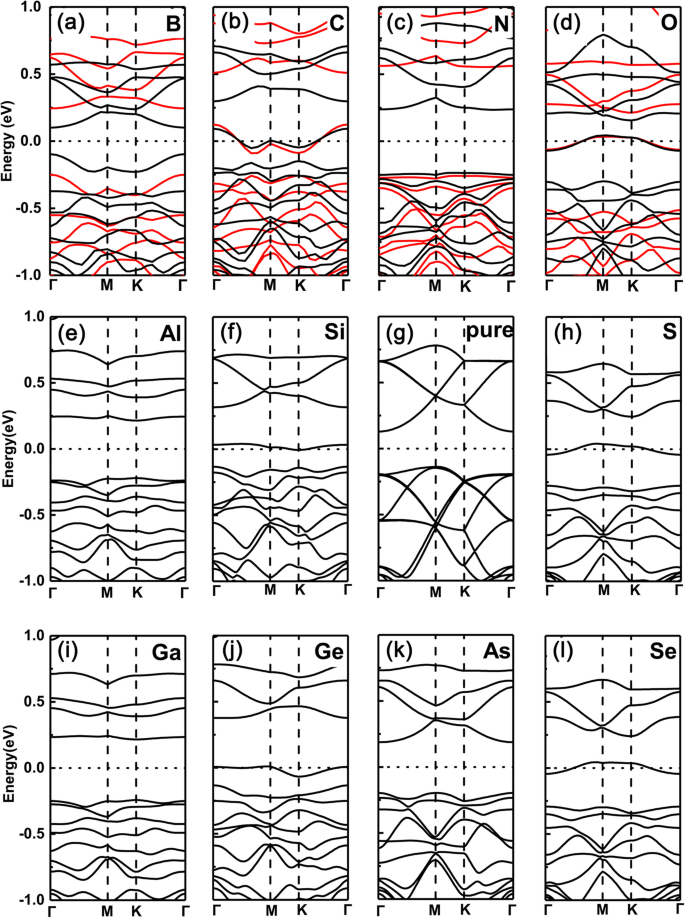
Bandstructuren van de verschillende doteermiddelen in GeP3 monolaag met vervangend P-atoom. een B, b C, c N, d O, e Al, f Si, g P, u S, ik Ga, j Pure GeP3 , k Als, l zie. Berekende bandstructuren voor een \( \mathsf{2}\times \mathsf{2} \) supercel met verschillende doteringen in GeP3 monolaag van groep III tot VI, met respectievelijk vervangende P-atomen, samen met die van zuiver GeP3 monolaag. Zowel de PBE- als de HSE06-functionaliteit worden gebruikt in de bovenste rij
Om de oscillaties van de overgang van halfgeleidende naar metallische eigenschappen beter te presenteren, hebben we de veranderende trend van de bandafstand uitgezet als de verschillende doteermiddelen, zoals respectievelijk getoond in Fig. 4a en b. Het is duidelijk dat we kunnen zien dat de overgang van halfgeleidende naar metallische eigenschappen drastisch omkeert. In het bijzonder treden metallische (halfgeleidende) halfgeleidende (metaalachtige) oscillaties op bij het vervangen van de Ge (P) -plaats als de doteermiddelen variërend van groep III tot VI. Daarnaast hebben we ook een interessant fenomeen gevonden dat aantoont dat de grootte van de bandgap bijna hetzelfde blijft als intrinsieke GeP3 monolaag wanneer de doteermiddelen dezelfde valentie-elektronen hebben als het Ge-atoom. Wanneer de doteermiddelen echter twee elektronen meer hebben dan het Ge-atoom, verandert de grootte van de bandafstanden relatief groter. Niettemin, voor de doteermiddelen op P-plaatsen, ongeacht het aantal valentie-elektronen, verandert de grootte van de bandhiaten relatief altijd groot. Dit kan worden begrepen door het gezamenlijke effect van de straal van het atoom en de beschikbare valentie-elektronen, namelijk doteringen met bijna dezelfde (kleinere of grotere) straal en valentie-elektronen als (dan) Ge-atoom veroorzaken een relatief kleiner (groter) effect op de elektronische eigenschappen, zoals de bandgap. Dit betekent dat men niet alleen de oscillaties van halfgeleidende-metaalovergangen kan afstemmen, maar ook de grootte van de bandgap kan afstemmen door de juiste doteermiddelen en de verschillende doteringsplaatsen te kiezen.
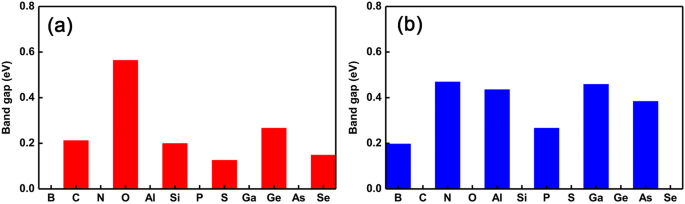
Band gaps van alle enkelvoudig gedoteerde systemen. Band gaps van gedoteerde GeP3 monolagen met de verschillende doteermiddelen variërend van groep V tot VI. een de vervanging van Ge-atomen en b de substitutie P-atomen, respectievelijk
Om de verandering van de elektronische structuren van verschillende doteermiddelen in de GeP3 . te begrijpen monolaag, hebben we de partiële dichtheid van toestanden (PDOS) van intrinsieke en dopinggroep IV-V in GeP3 uitgezet monolaag, zoals respectievelijk getoond in Fig. 5a-d. Het is duidelijk te zien dat de maximale valentieband (VBM) en de minimale geleidingsband (CBM) van de GeP3 monolaag komt voornamelijk voort uit zowel p-orbitalen van Ge- als P-atomen. Wanneer de doteermiddelen met hetzelfde aantal valentie-elektronen als het Ge-atoom, zoals C en Si, beschikbaar zijn, zullen er onzuiverheden zijn die zich net boven de VBM van intrinsiek GeP3 bevinden monolaag omdat het p-orbitale energieniveau van C en Si hoger is dan dat van het P-atoom (zie figuur 5a). Daarom is de geleidende eigenschap intact en is de grootte van de verandering in de bandafstand relatief klein. Wanneer de doteermiddelen echter één elektron meer hebben dan het Ge-atoom, zoals N, P en As, zullen er ook onzuiverheidstoestanden in de bandgap zijn en de onzuiverheidstoestanden komen voort uit de hybridisatie van splitsing van CBM (dominant) en de toestanden van doteermiddelen (zie Fig. 5b).
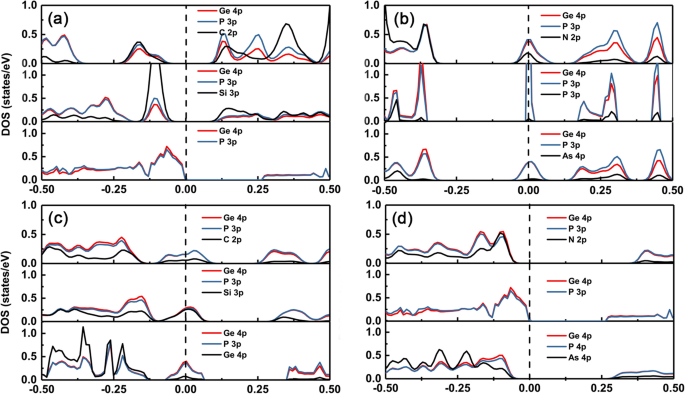
DOS voor de gedoteerde systemen. De partiële toestandsdichtheid (rechts) voor groep IV (C, Si en Ge) en groep V (N, P en As) atomen gedoteerd GeP3 . De verticale zwarte stippellijn is het Fermi-niveau. (a) en (b) gesubstitueerd Ge-atoom, (c) en (d) gesubstitueerd P-atoom
Integendeel, voor de dotering op de P-site, wanneer de doteermiddelen één valentie-elektron minder hebben dan het P-atoom zoals groep IV, zullen er onzuiverheidstoestanden zijn over het Fermi-niveau, en de onzuiverheidstoestanden zijn samengesteld uit het splitsen van VBM (dominante ) en de toestanden van doteermiddelen. Terwijl, wanneer de doteermiddelen hetzelfde aantal valentie-elektronen hebben als het P-atoom, zoals groep V, de gedoteerde systemen nog steeds een halfgeleidende karakteristiek behouden (zie figuur 5c). De bandgaps worden relatief groter dan die van intrinsieke GeP3 monolaag vanwege de grotere mismatch van roosterconstanten. Bovendien hebben we ook waargenomen dat de bandafstand van de N-doteringsstof groter is dan die van de As-doteringsstof bij het vervangen van P-atomen. Dit komt omdat het p-orbitale energieniveau van het As-atoom hoger is dan dat van het N-atoom, dus hoe hoger het energieniveau p-orbitaal, hoe meer de opwaartse verschuiving van onzuiverheidstoestanden weg van VBM (zie figuur 5d).
Logische relaties voor co-dopingsystemen
Op basis van de bovengenoemde bevindingen van enkele verschillende doteringsmiddelen kunnen we daarom co-dopingsystemen ontwerpen om te voldoen aan de elektronische eigenschappen die we willen. Hier tonen we alleen de resultaten van B, C, N en O als voorbeelden om het co-doping-effect te illustreren, maar de conclusie is robuust tegen de verschillende geselecteerde doteermiddelen. Bijvoorbeeld, op de Ge-site co-doping kunnen zowel de twee doteringen met één valentie-elektron minder natuurlijk leiden tot halfgeleidende eigenschappen, terwijl voor de twee doteerstoffen met een steeds minder aantal valentie-elektronen, de co-dopingsystemen dus ook kunnen een halfgeleidende eigenschap hebben.
Voor de twee doteermiddelen met één minder (meer) en hetzelfde valentie-elektron, behouden de co-dopingsystemen echter nog steeds de metallische eigenschap als een minder (meer) aantal valentie-elektron-doteermiddelen resulterende eigenschap. Vereenvoudig, dit idee wordt precies bevestigd door onze verdere dichtheidsfunctionaaltheorie (DFT) -berekeningen van co-gedoteerde systemen, zie de resultaten in Fig. 6a-l voor de bandstructuren van B, C, N en O co-gedoteerde GeP3 monolaag.
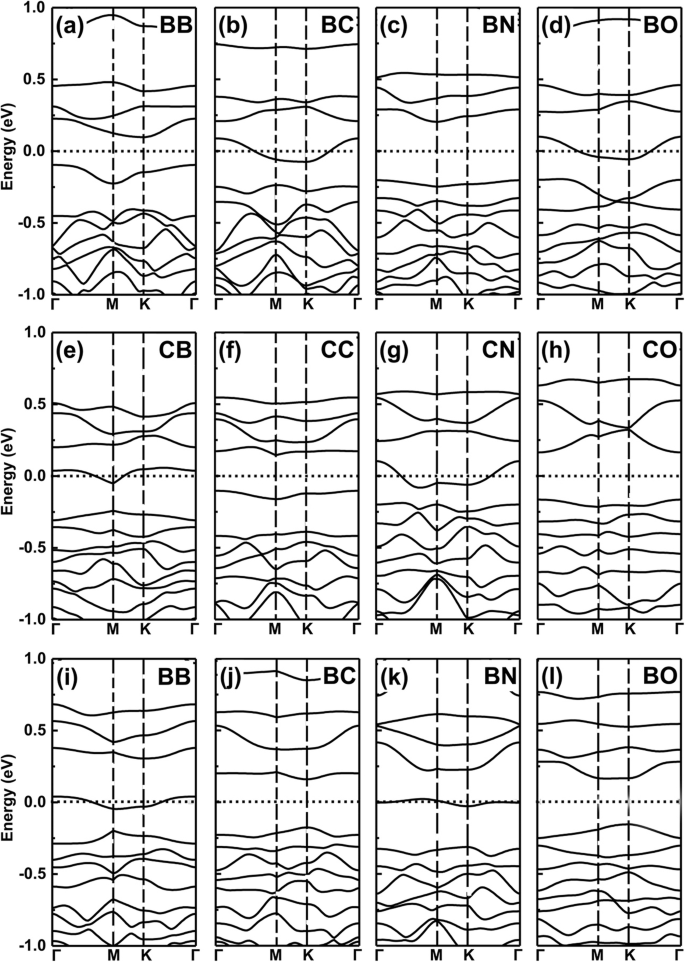
Bandstructuren van co-gedoteerde systemen. De bandstructuren van B, C, N en O samen gedoteerd GeP3 monolaag. een –d De twee doteerstoffen vervangen twee Ge-atomen in GeP3 monolaag, e –u de twee doteerstoffen vervangen twee P-atomen, i –ik de twee doteerstoffen vervangen respectievelijk één Ge-atoom en één P-atoom
Nu kunnen we een voorbeeld van een logisch-achtige bewerking "AND" geven, waarbij de metaaleigenschap wordt ingesteld als "M" en de halfgeleidende karakteristiek als "S". We definiëren de logische relaties:respectievelijk M AND M = S, S AND S = S, en M AND S = M. Hierin gehoorzamen deze bevindingen die we hierboven hebben verkregen aan dergelijke logisch-achtige relaties, bijvoorbeeld de doteermiddelen met één meer en één minder valentie-elektron resulteren in metaaleigenschap, maar wanneer we de twee doteermiddelen als co-doping gebruiken, zoals B en N op Ge-sites zoals getoond in Fig. 7a en b, worden de co-gedoteerde systemen zoals verwacht halfgeleidende eigenschappen, zie Fig. 6c. Als we kiezen voor BC co-gedoteerde GeP3 monolaagsysteem, vertoont het een metaalachtig kenmerk dat het geval is bij M AND S (zie Fig. 4a, b). Hetzelfde voor CN-, N-O- en B-O-atomen co-doping in GeP3, vervanging van twee Ge-atomen, twee P-atomen of één Ge- en P-atoom, zoals respectievelijk getoond in Fig. 7c-f.
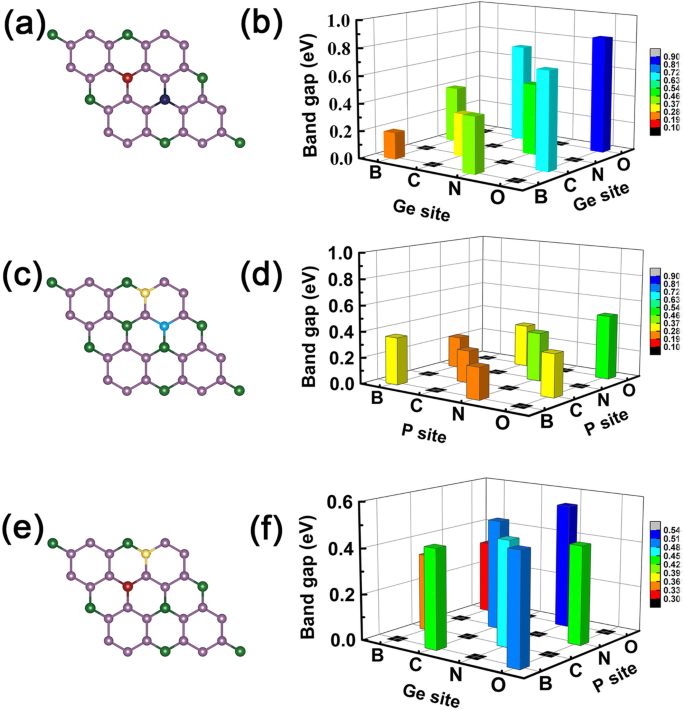
Band gaps van alle co-gedoteerde systemen. De omvang van de band gaps van gecodopeerd GeP3 monolaag, de linkers zijn de schets van co-gedoteerde sites, en de rechten zijn de grootte van bandhiaten die overeenkomen met de doping-elementen. een , b Het geval voor doping-elementen bezetten de twee Ge-atomen. c , d Het geval voor doping-elementen bezetten de twee P-atomen. e , v Het geval voor doping-elementen bezetten respectievelijk de Ge- en P-atomen
Ten slotte hebben we de stabiliteit van zowel de enkelvoudig gedoteerde systemen als de co-gedoteerde systemen gecontroleerd om er zeker van te zijn of ze verder in het experiment kunnen worden gerealiseerd. De vormingsenergie wordt berekend met behulp van de Vgl. (1) en (2) voor respectievelijk single-doping- en co-dopingzaken. De resultaten zijn aanwezig in Fig. 8a-e. Uit Fig. 8a en b kunnen we duidelijk zien dat de E f van de enkele doteermiddelen in Ge-sites zijn allemaal dicht bij die van GeP3 monolaag (instelling op nul als referentiepunt), met uitzondering van C-, N- en S-atomen. We hebben ook opgemerkt dat, voor doteerstoffen van B-, O-, P-, Ge- en Se-atomen, de vormingsenergieën veel kleiner zijn dan die van andere doteerstoffen, wat aangeeft dat ze heel gemakkelijk te doteren zijn in een experiment. Voor doteerstoffen in de P-plaatsen hebben doteerstoffen van B-, O-, P- en Ge-atomen een relatief kleinere vormingsenergie en zijn ze ook gemakkelijk te doteren. C, N, Al en Ga zijn niet gemakkelijk voor doping.
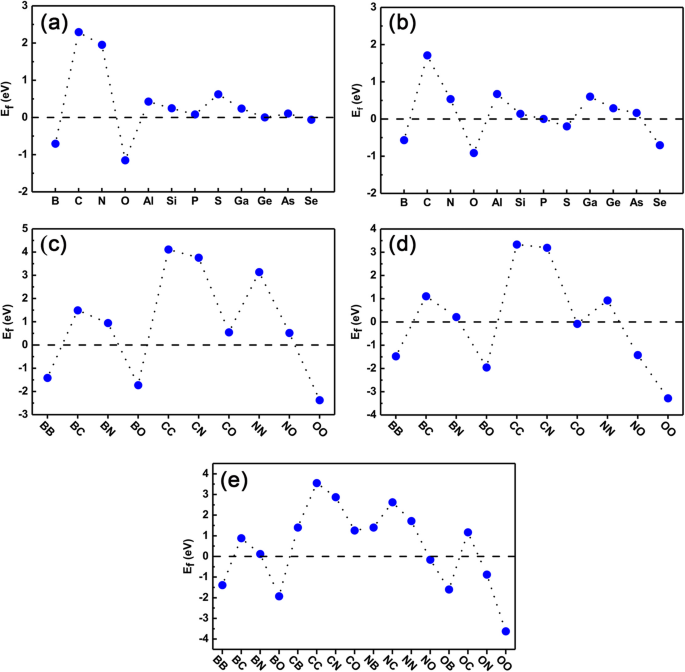
Vormingsenergie van alle gedoteerde systemen. De berekende vormingsenergie van enkelvoudige doping- en co-dopingsystemen. een , b zijn respectievelijk het door doteringsmiddel gesubstitueerde Ge-atoom en P-atoom; c –e De co-doteermiddelen gesubstitueerd door respectievelijk twee Ge-atomen, twee P-atomen en één Ge-atoom en één P-atoom
Wat betreft de vormingsenergie van co-doping, zijn Fig. 8c-e de vormingsenergie van co-doping waarbij twee doteringen de posities innemen van twee Ge-atomen (aangeduid als Ge-Ge-plaatsen), twee P-atomen (aangeduid als PP plaatsen), respectievelijk één Ge- en één P-atoom (aangeduid als Ge-P-plaatsen). Hier tonen we alleen de resultaten van doteermiddelen van B, C, N en O als voorbeeld. Voor Ge-Ge-sites en PP-sites kan de vormingsenergie van co-doping ruwweg worden geschat door de vormingsenergieën van doping met één element afzonderlijk te middelen. Het is duidelijk dat voor BB-, BO- en OO-co-doping in Ge-Ge-sites en BB-, BO-, NO- en OO-co-doping in PP-sites de vormingsenergieën relatief klein zijn en gemakkelijk in het experiment kunnen worden gerealiseerd. Voor CC-, CN- en NN-co-doping in Ge-Ge-sites en CC- en CN-co-doping in PP-sites zijn de vormingsenergieën echter relatief groter, wat aangeeft dat ze moeilijk te dopen zijn in experiment. Voor co-doping van Ge-P-sites, zoals getoond in Fig. 8e, wordt de vormingsenergie complexer dan co-doping van Ge-Ge of PP-sites omdat er ladingsoverdracht tussen de doteermiddelen is. In elk geval hebben de co-doping van BB, BO en OO de kleinere vormingsenergieën, terwijl co-doping CC, CN en NN grotere vormingsenergieën hebben. In het algemeen hangt de vormingsenergie sterk af van het aantal valentie-elektronen van het doteringsmiddel. In het bijzonder, wanneer de twee doteermiddelen met één (meer) elektron minder dan de gesubstitueerde atomen, is de vormingsenergie van het co-gedoteerde systeem lager (hoger) dan die van de overeenkomstige enkelvoudige doteringen zoals BB (NN) co-gedoteerde Ge-plaatsen. Dit komt omdat er concurrentie bestaat tussen de verminderde (verhoogde) energie van verminderde (verhoogde) elektronen van gedoteerde systemen en Coulomb-afstoting. Voor de hole-hole co-doping is de energie van het eerste geval veel groter dan het laatste geval, wat resulteert in de vrij verminderde vormingsenergie in co-dopingsystemen zoals BB, terwijl voor de elektron-elektron co-doping beide de eerste en latere gevallen leiden tot de hogere vormingsenergie zoals NN. Voor gaten- en elektronen-co-gedoteerde systemen zoals BN-co-gedoteerde Ge-locaties is de vormingsenergie echter dramatisch lager dan de overeenkomstige enkel-gedoteerde gevallen. Dit komt omdat in een dergelijk co-gedoteerd systeem er geen energiewinst is van netto toegevoegde of verminderde elektronen in de systemen, en de Coulomb-interactie een beslissende rol speelt bij de vorming van co-gedoteerde doteermiddelen. Al met al, als we onze eerdere studies van elementdoping in zwart fosforeen samenvatten, moet erop worden gewezen dat onze huidige studies een zekere mate van universaliteit hebben en worden verwacht voor het toepassen van andere 2D-halfgeleidermonolagen, zoals BN, MoS2 , enzovoort.
Ten slotte, om de stabiliteit van de bovengenoemde gedoteerde systemen te controleren in vergelijking met het niet-gedoteerde geval, hebben we de AIMD (ab initio moleculaire dynamica) uitgevoerd om de energie versus tijd te tonen, zoals getoond in Fig. 9a-f. We kunnen duidelijk zien dat de oscillerende amplitude convergent zal zijn zolang de tijd lang genoeg duurt (~ 4 ps), wat impliceert dat de gedoteerde systemen niet zullen instorten tegen thermische fluctuaties tot 300 K voor C-gedoteerde GeP3 in afb. 9a. Zelfs voor de meest actieve met C-atoom gedoteerde GeP3 , kan de extreme temperatuur oplopen tot 300 K, zoals weergegeven in Fig. 9c. Bovendien nemen we ook metaal Al-substitutie op de Ge-site als voorbeeld, het berekende resultaat wordt getoond in Fig. 9e en f, waaruit we duidelijk kunnen zien dat de energie-oscillatie-amplitude geleidelijk afneemt met de tijdsduur, wat betekent dat de energie zou kunnen convergent zijn zolang de tijd lang genoeg duurt en de structuur van het gedoteerde systeem thermisch stabiel is tegen thermische fluctuaties. Daarom kunnen we verwachten dat dergelijke gedoteerde systemen kunnen worden gerealiseerd in verdere experimenten gezien de hoogwaardige GeP3 monolaag wordt voorbereid.
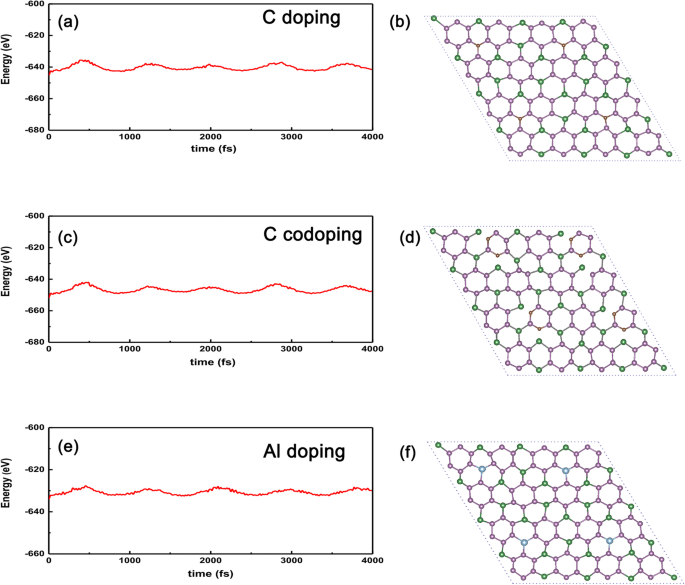
AIMD voor de C-, twee C- en Al-atoom-gedoteerde systemen. AIMD bevestigt de thermische stabiliteit van a Met atoom gedoteerde GeP3 met vervanging van Ge-atoom, c twee C-atomen gedoteerd GeP3 met vervanging van twee P-atomen, en e Al-atoom-gedoteerde GeP3 met vervanging bij 300 K. De structuren in b C, d twee C-atomen en f Al komen overeen met hun uiteindelijke structuren na 4000 fs
Ten slotte willen we de betrouwbaarheid van ons hier gepresenteerde onderzoek bespreken. Onze conclusies die hier worden gepresenteerd, zijn theoretisch voorspelde resultaten, maar zijn zeer betrouwbaar. Dit komt omdat ons gastheermateriaal dat hier wordt gebruikt is gerapporteerd en de bulkfase van gelaagde GeP3 bestaat al [26]. Dus onze bestudeerde doping-geïnduceerde gerelateerde fenomenen moeten verder worden bevestigd in het experiment zodra monolaag GeP3 verder wordt gerealiseerd. Vervolgens zou het doteren van de overeenkomstige atomen kunnen worden uitgevoerd. Voor de eenvoud doping elektron of gat in monolaag GeP3 kan worden gerealiseerd door de adsorptie van sommige moleculen.
Conclusie
Samenvattend hebben we de elektronische eigenschappen van groep III tot VI doteringen onderzocht in 2D GeP3 monolaag en vind dat de gedoteerde GeP3 met substitutie op de Ge-site vertoont metaal-halfgeleideroscillaties als functie van het aantal valentie-elektronen van doteermiddelen, terwijl dergelijke oscillaties worden omgekeerd met substitutie op de P-site. Op basis van de resultaten van enkelvoudige doteringen zouden we de geleidende eigenschappen van co-doping in GeP3 kunnen voorstellen. , die kan worden verkregen door een eenvoudige logische bewerking. Ten slotte berekenen we de vormingsenergieën van verschillende doteringen en ontdekken dat sommige van de co-gedoteerde systemen, vooral voor de co-doping van elektronen en gaten, energetisch gunstiger zijn vanwege de Coulomb-aantrekking. Onze bevindingen presenteren niet alleen een nieuw fenomeen, maar stellen ook een intrigerende route voor om de elektronische eigenschappen in 2D binaire halfgeleiders af te stemmen.
Beschikbaarheid van gegevens en materialen
De datasets die zijn gegenereerd tijdens en/of geanalyseerd tijdens het huidige onderzoek zijn op aanvraag verkrijgbaar bij de corresponderende auteur.
Afkortingen
- 1D:
-
Eendimensionaal
- 2D:
-
Tweedimensionaal
- AIMD:
-
Ab initio moleculaire dynamica
- BP:
-
Zwart fosforeen
- CBM:
-
Minimaal geleidingsband
- DFT:
-
Dichtheidsfunctionaaltheorie
- HSE06:
-
Hybride dichtheid functioneel
- PBE:
-
Perdew-Burke-Ernzerhof
- PDOS:
-
Gedeeltelijke dichtheid van toestanden
- VBM:
-
Maximale valentieband
Nanomaterialen
- Valentie en kristalstructuur
- Elektronen en "gaten''
- Adsorberende verwijdering van koper(II)-ionen uit waterige oplossing met behulp van een magnetiet nano-adsorbens uit afval van walshuid:synthese, karakterisering, adsorptie en kinetische modellering O…
- Bereiding van palladium(II)-ion-imprinted polymere nanosferen en de verwijdering van palladium(II) uit waterige oplossing
- Synthese en CO-oxidatieactiviteit van 1D gemengd binair oxide CeO2-LaO x ondersteunde gouden katalysatoren
- 5-aminolevulinezuur-squaleen nanoassemblages voor tumorfotodetectie en therapie:in vitro studies
- Synthese en karakterisering van gemodificeerde BiOCl en hun toepassing bij adsorptie van kleurstoffen met een lage concentratie uit een waterige oplossing
- Morfologie, structuur en optische eigenschappen van halfgeleiderfilms met GeSiSn-nano-eilanden en gespannen lagen
- Multiband- en breedbandabsorptieverbetering van monolaag grafeen bij optische frequenties van meerdere magnetische dipoolresonanties in metamaterialen
- Alkalimetaal-geadsorbeerde g-GaN-monolaag:ultralage werkfuncties en optische eigenschappen
- Uitstekende lichtopsluiting van hemiellipsoid- en geïnverteerde hemiellipsoid-gemodificeerde halfgeleider nanodraadarrays